DCA for SARS-CoV-2
Epistatic models predict mutable sites in SARS-CoV-2 proteins and epitopes
Epistatic models predict mutable sites in SARS-CoV-2 proteins and epitopes
For each site of the SARS-CoV-2 Wuhan-Hu-1 proteome (Accession NC_045512) included in a PFAM domain, we introduce a Direct Coupling Analysis (DCA) mutability score to predict mutable and constrained sites.
Sites of the SARS-Cov-2 Wuhan-Hu-1 proteome with high DCA-Mutability Scores are more likely to mutate. We validate our mutability predictions with the mutations observed in SARS-CoV-2 proteomes deposited in the GISAID database.
Paper: Epistatic models predict mutable sites in SARS-CoV-2 proteins and epitopes Rodriguez-Rivas, Giancarlo Croce, Maureen Muscat, Martin Weigt, PNAS January 25, 2022 119 (4) e2113118119; https://doi.org/10.1073/pnas.2113118119.
Run the Jupyter-notebook dca_sarscov2.ipynb on Google Colab at this link to reproduce key results from the paper and guide the data analysis. Colab allows you to execute the Python code through your browser (no need to clone the GitHub repository on your local machine).
The code for training sequence-based models to predict mutability scores is available at https://github.com/juan-rodriguez-rivas/covmut.
The results of our analysis are also available in the ./data folder on the Github page
The data structure is:
protein domain position_protein position_domain aa_Wuhan-Hu-1 mutability_score(IND) mutability_score(DCA) observed_mut_May2021 observed_mut_Dec2020 observed_mut_Jul2020
Spike bCoV_S1_RBD 349 1.0 S -1.3818 -1.2046 0.0 0.0 0.0
Spike bCoV_S1_RBD 350 2.0 V -1.9788 -1.1667 3.0 0.0 0.0
Spike bCoV_S1_RBD 351 3.0 Y -1.8017 -1.2678 8.0 1.0 1.0
Spike bCoV_S1_RBD 352 4.0 A -1.0140 -1.1347 23.0 3.0 0.0
We combine our DCA-Mutability Score with the IEDB site response frequency (IEDB-Response Frequency). This allows us to identify mutable (high DCA-Mutability Score) and immunologically relevant sites (high IEDB - Response Frequency). Amino acid substitutions at high-RF sites have a higher risk of inducing immune escape as many positively responding B and T cell epitopes are modified.
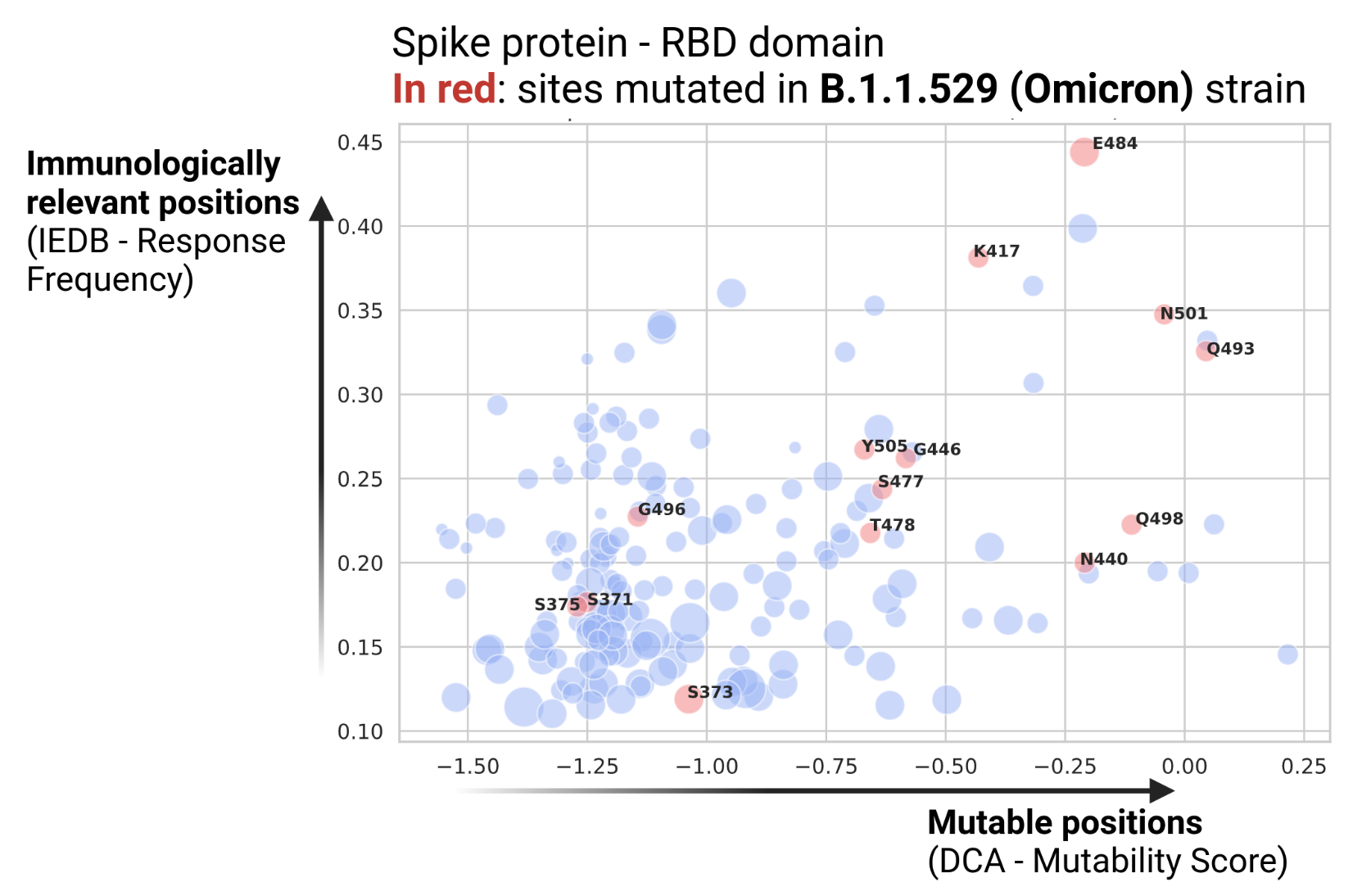
A key advantage of our data-driven modeling approach is the possibility to obtain predictions for all the protein domains in the SARS-CoV-2 proteome. Run the Jupyter-notebook to extend the DCA predictions to all 39 protein domains covering 81% of the entire proteome (8037 out of 9748 positions), and combine them with immunological IEDB data.